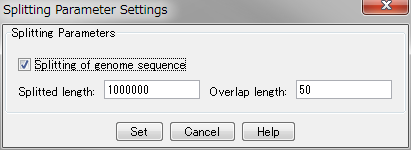
You are here: Home  Forum in silico Forum GenomeTraveler Questions (GT) GT: What is the meaning of the two values of "Splitting Option" in the fragment mapping?
Forum in silico Forum GenomeTraveler Questions (GT) GT: What is the meaning of the two values of "Splitting Option" in the fragment mapping?
|
Welcome,
Guest
|
TOPIC:

Time to create page: 0.039 seconds