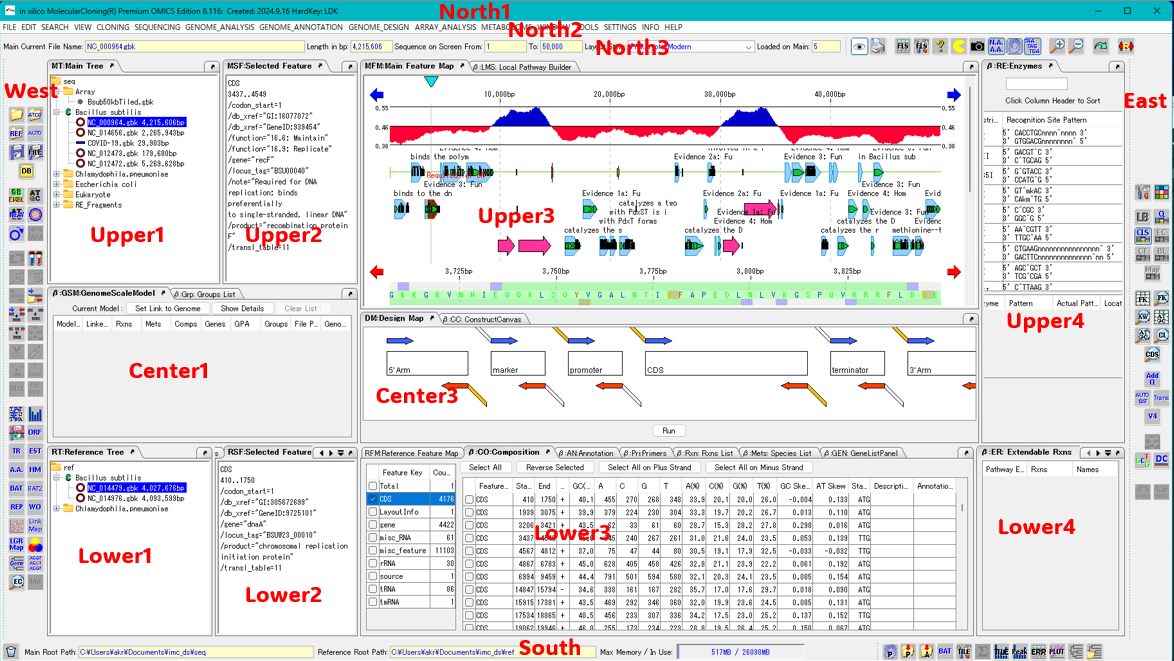
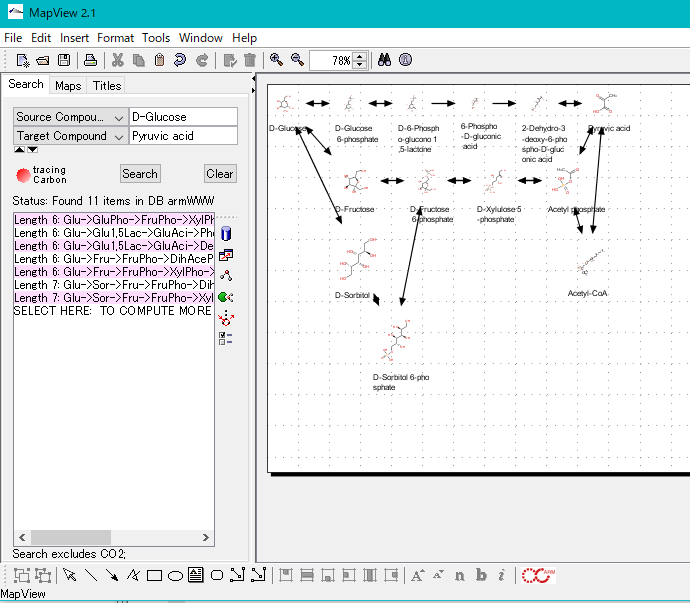
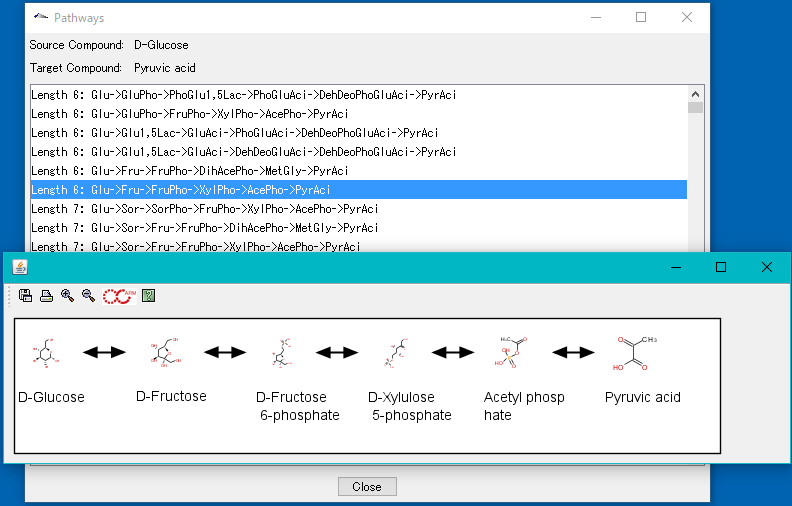
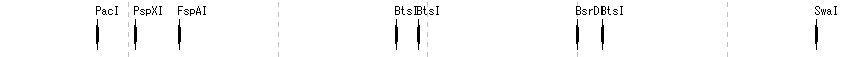サイト内全文検索
サイト内ポピュラータグ
trouble
5
SNP
6
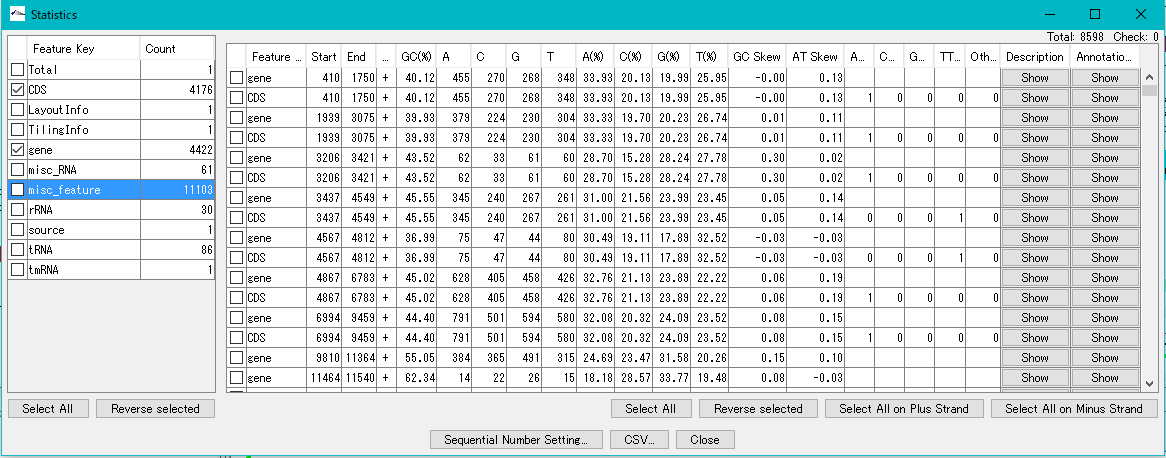
Qualifier
10
navigation
4
Mutation
5
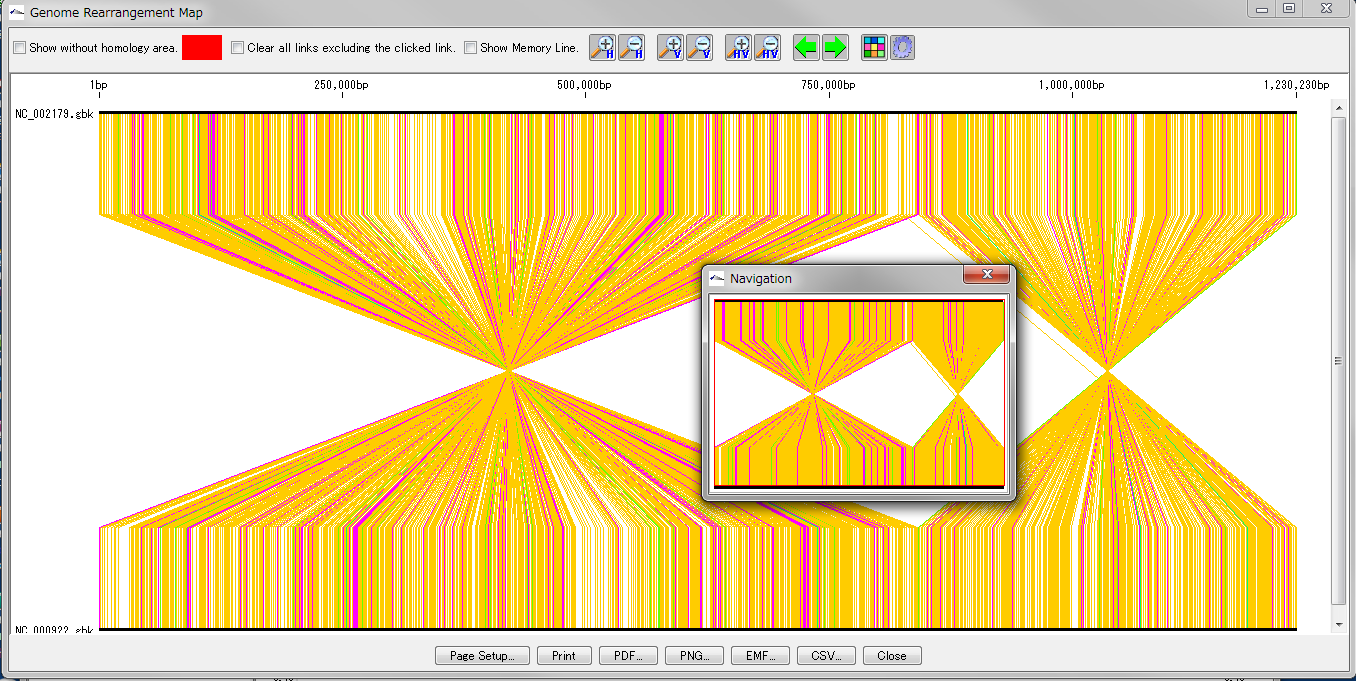
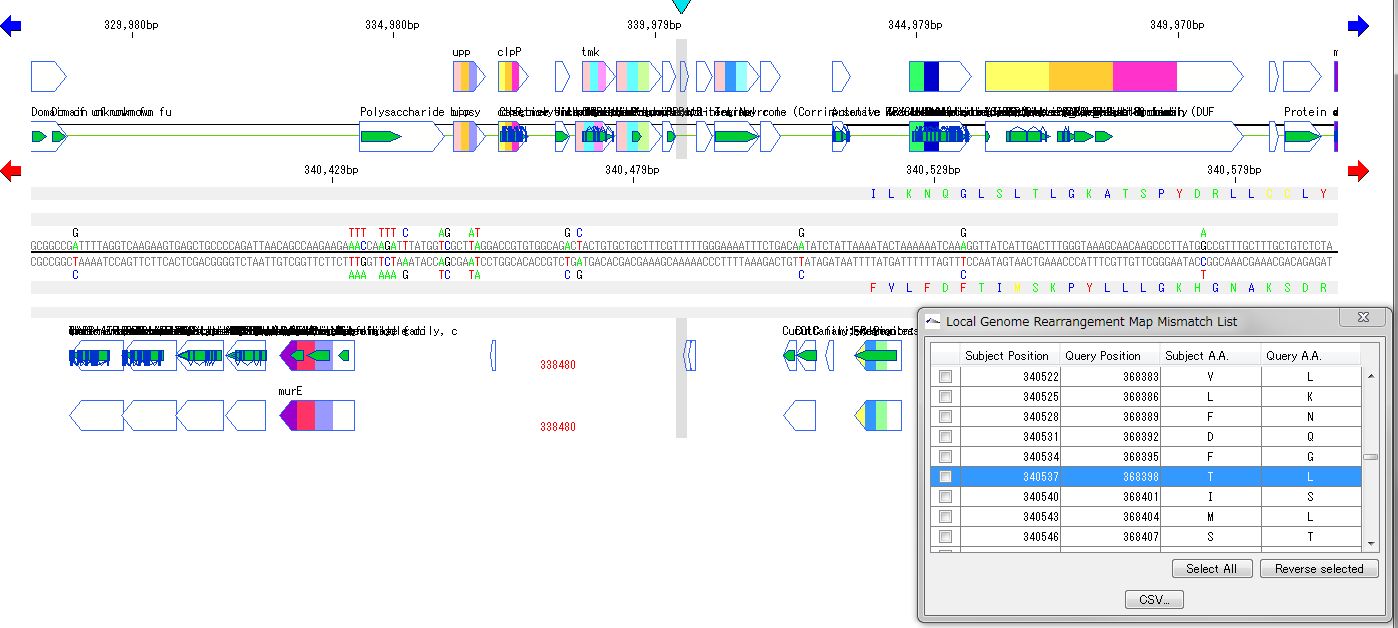
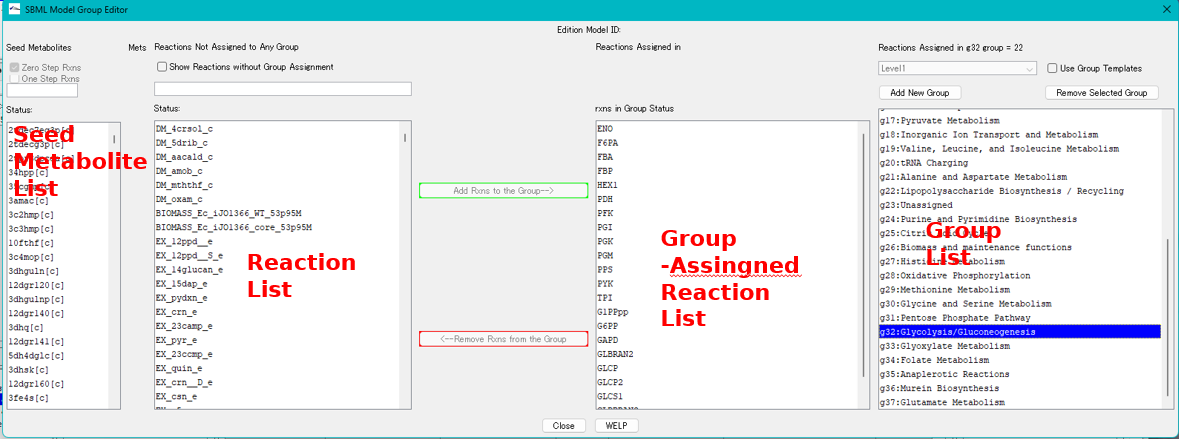
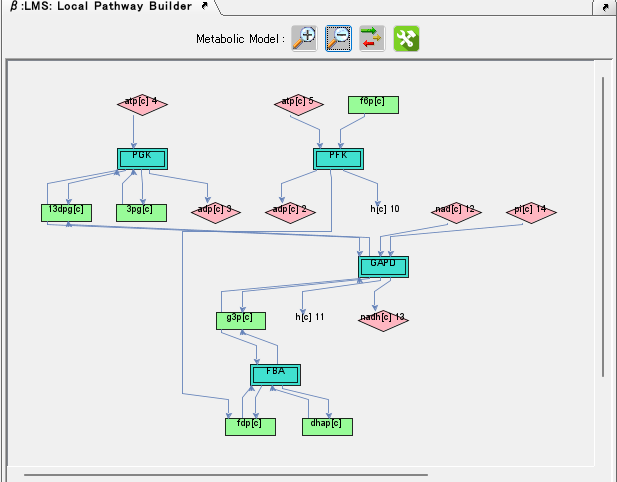
Local Genome Rearrangement Map
6
Linear Map
9
License
5
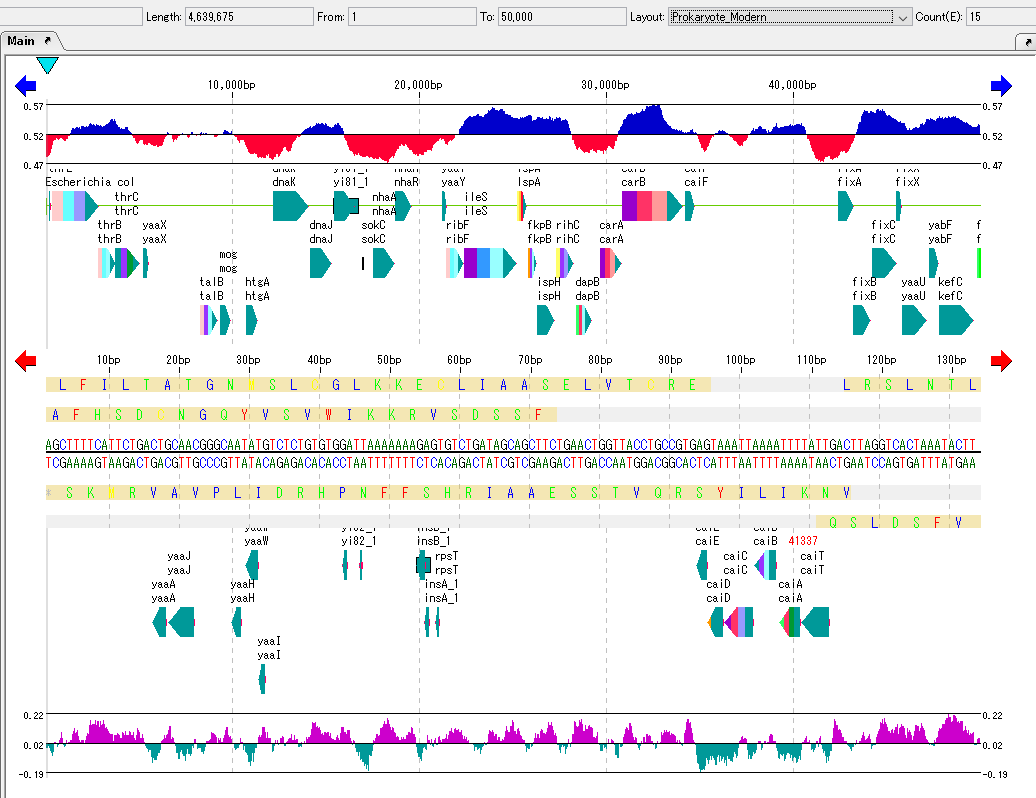
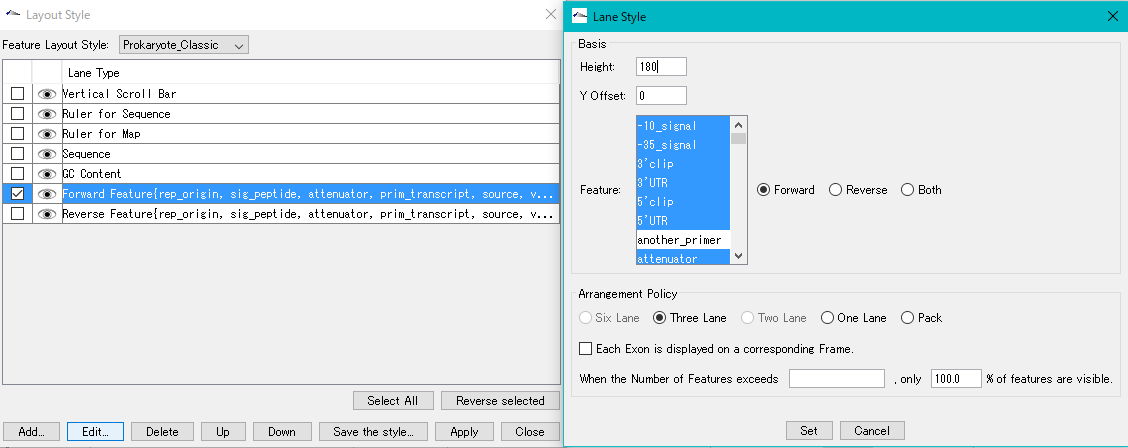
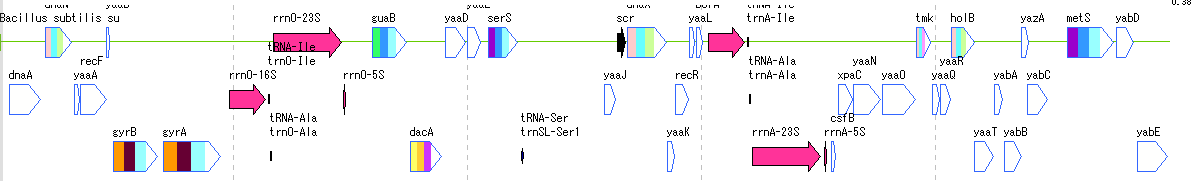
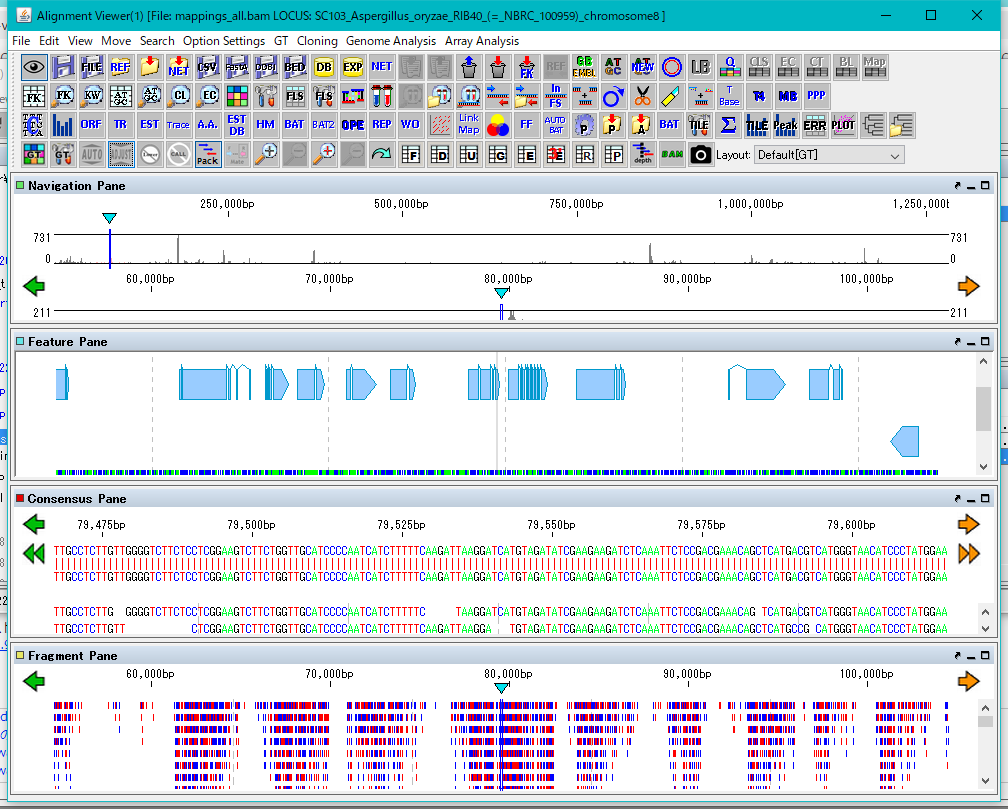
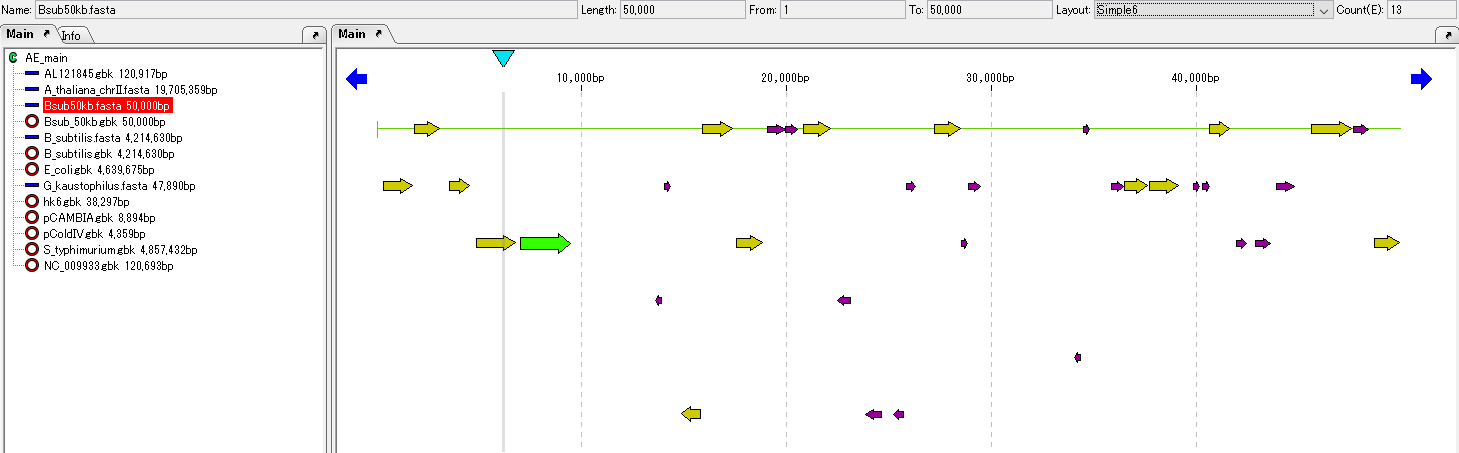
Lane
9
Genome Design
7
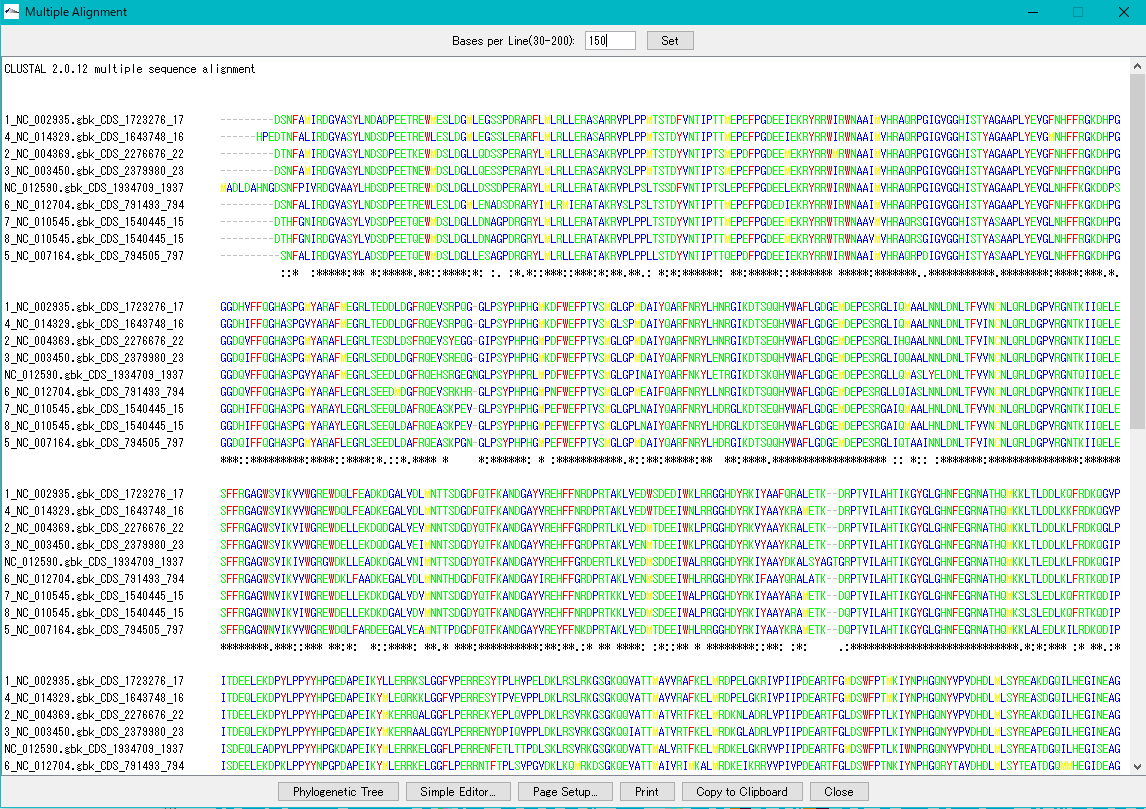
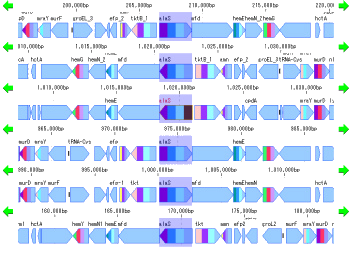
Gene Cluster Alignment
8
Frame
6
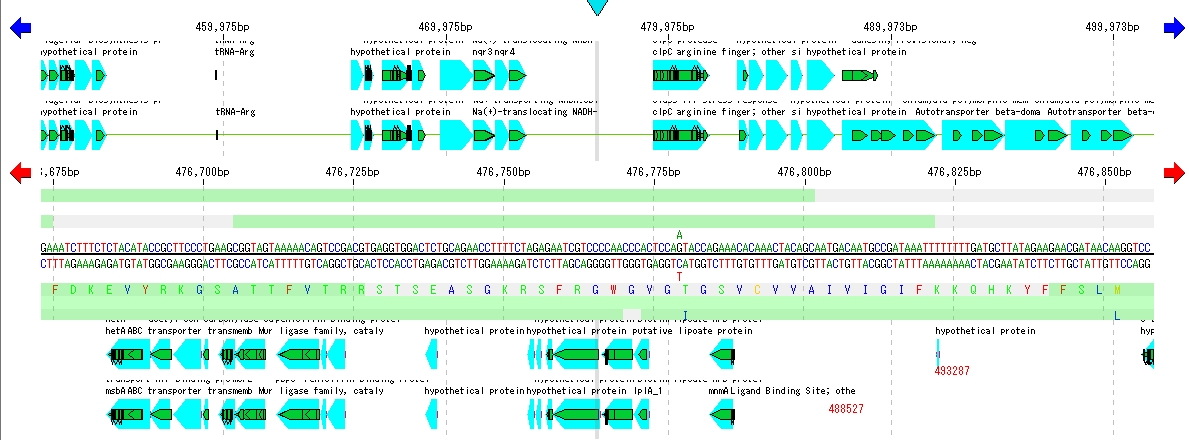
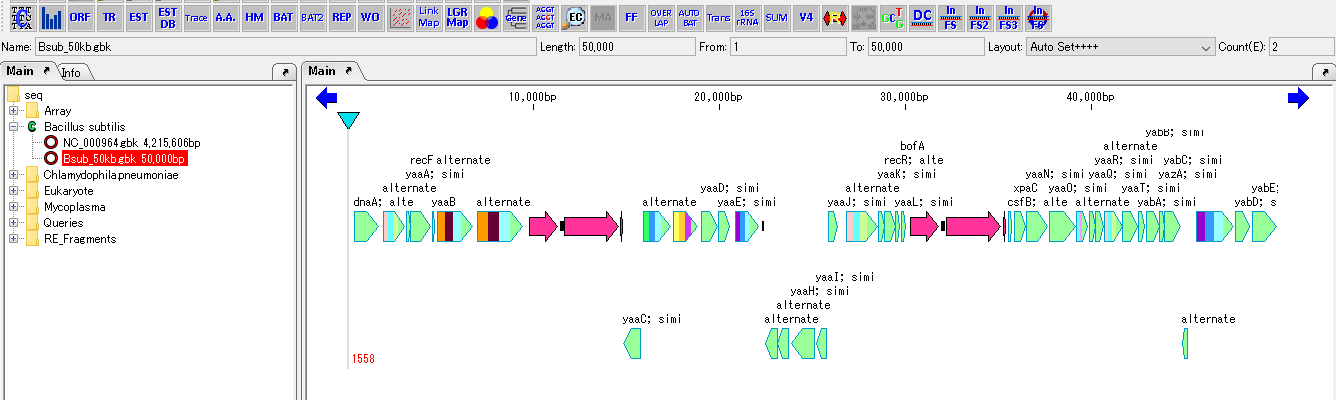
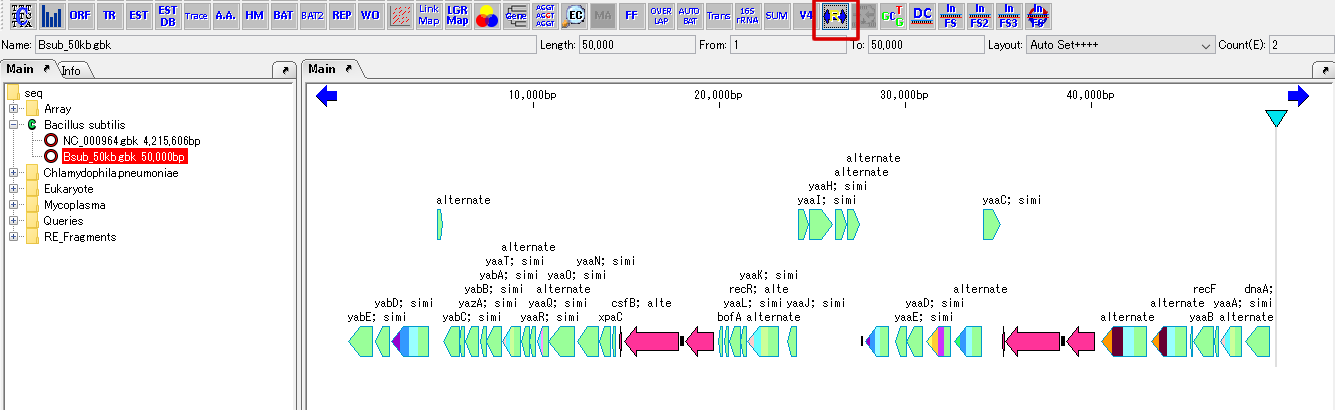
Feature Map
6
Feature Layout Style
5
feature key
5
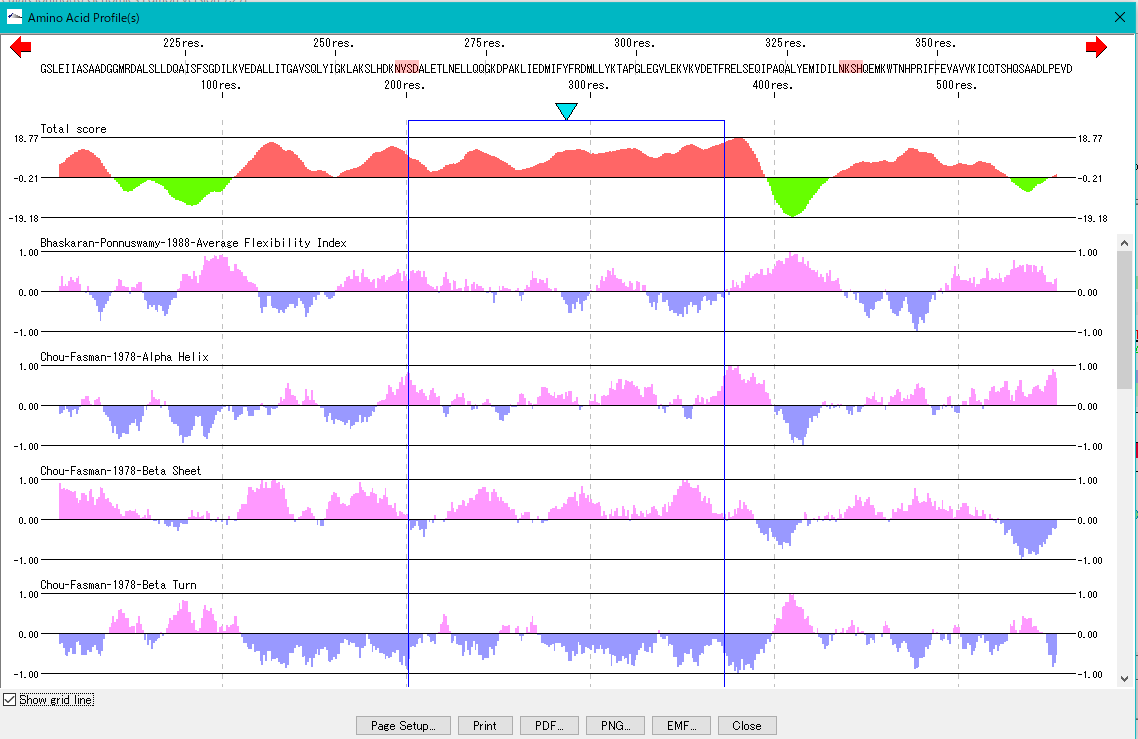
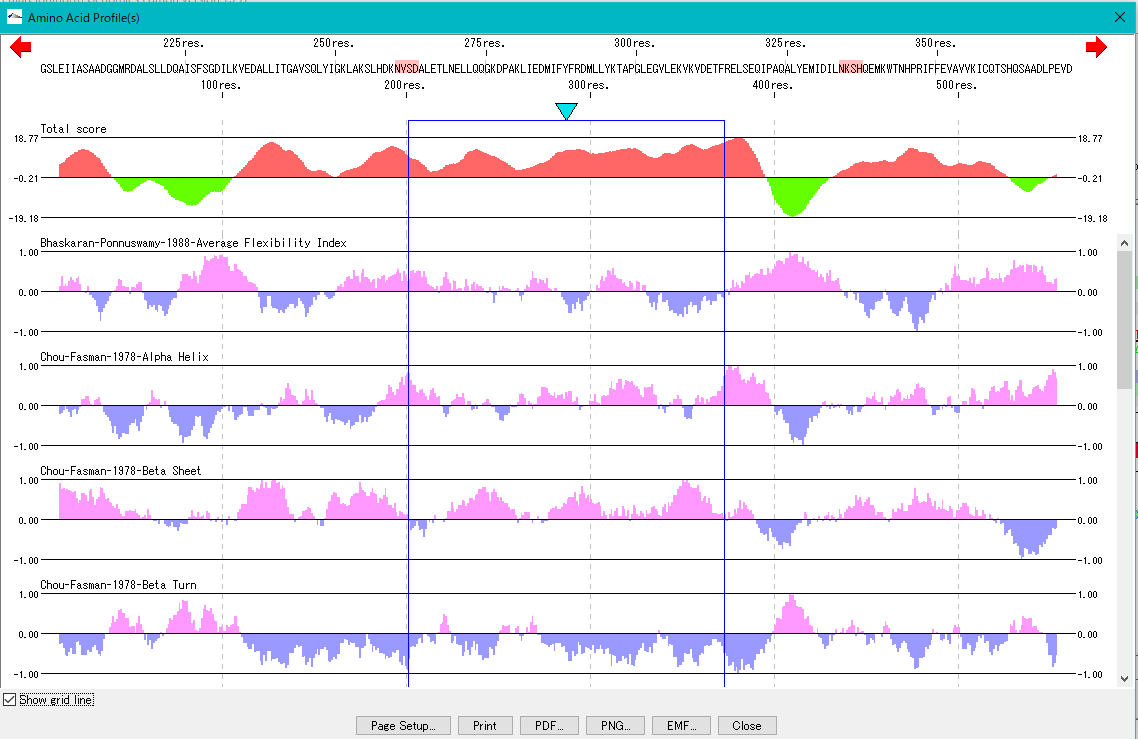
Feature
6
download
4
Dongle
4
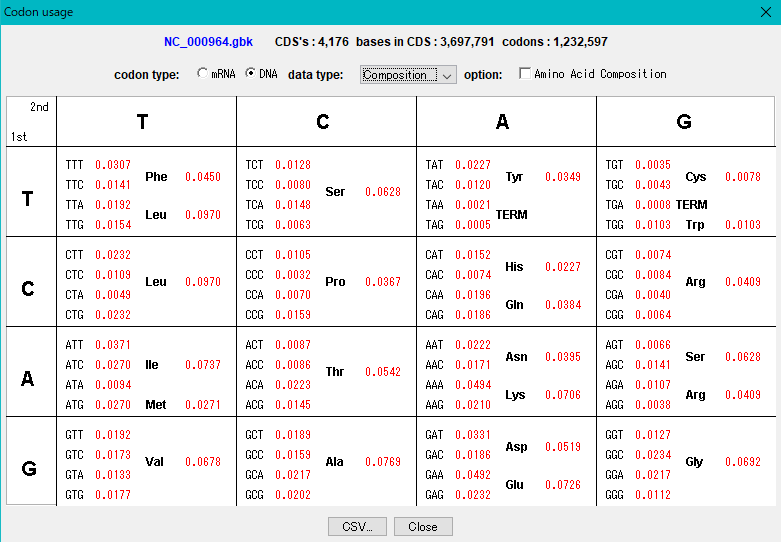
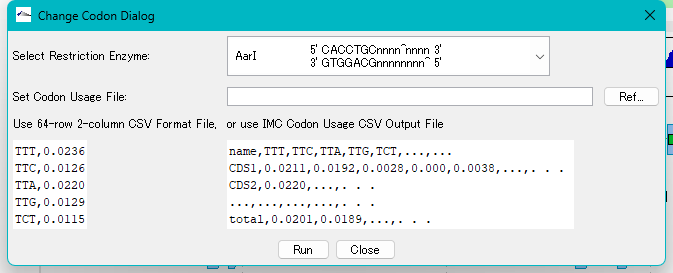
Codon
4
BLAST
11
 ドングルライセンス(HWキー)
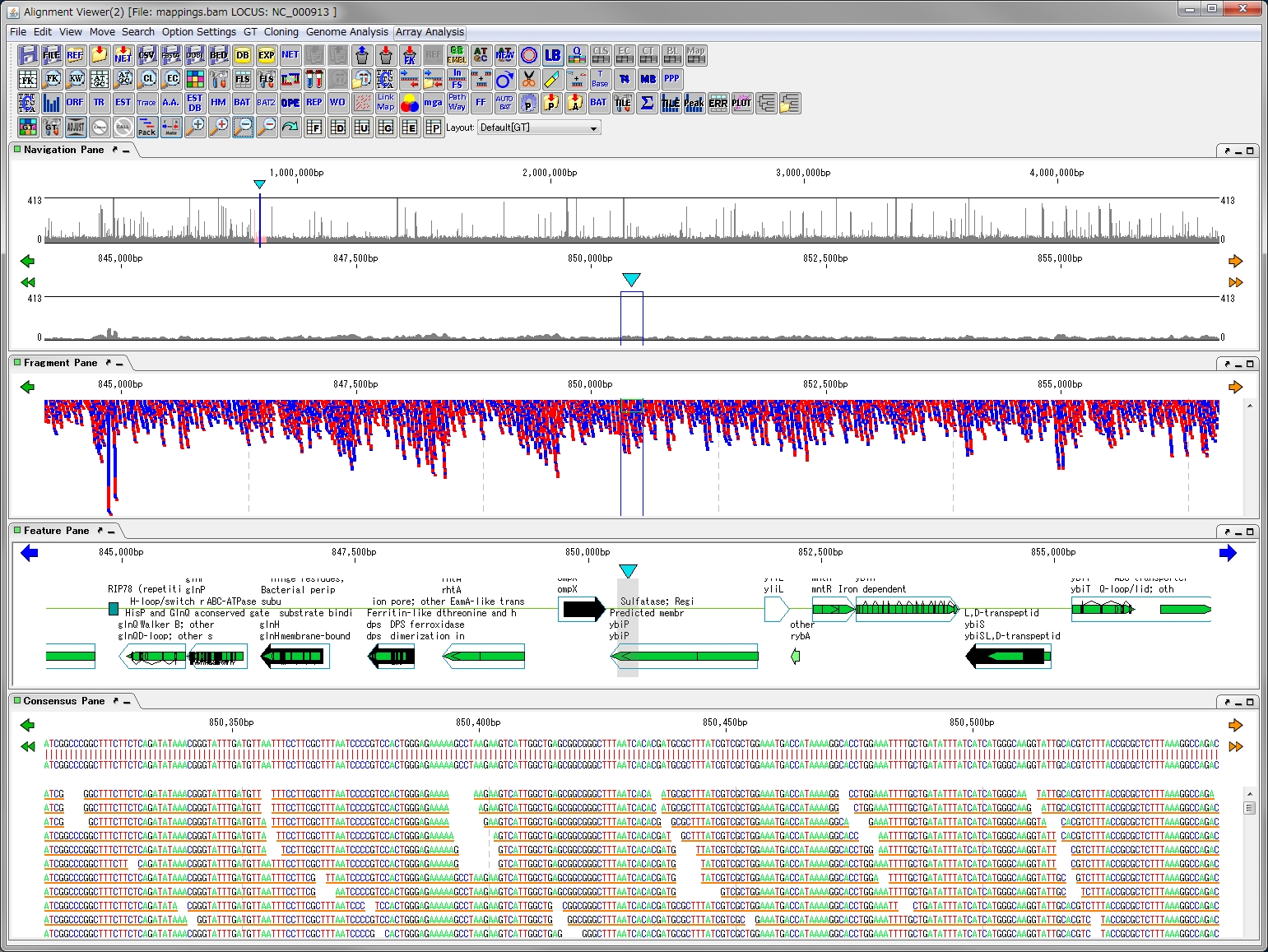
ドングルライセンス(HWキー) フィーチャーマップとレーン
フィーチャーマップとレーン 配列・データ入出力
配列・データ入出力 GenBank EMBLビューワ
GenBank EMBLビューワ 配列ビューワ
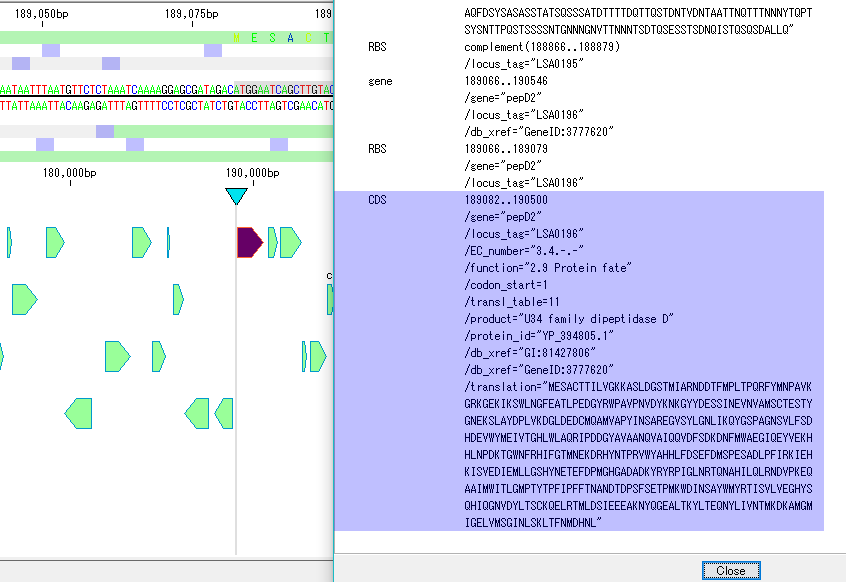
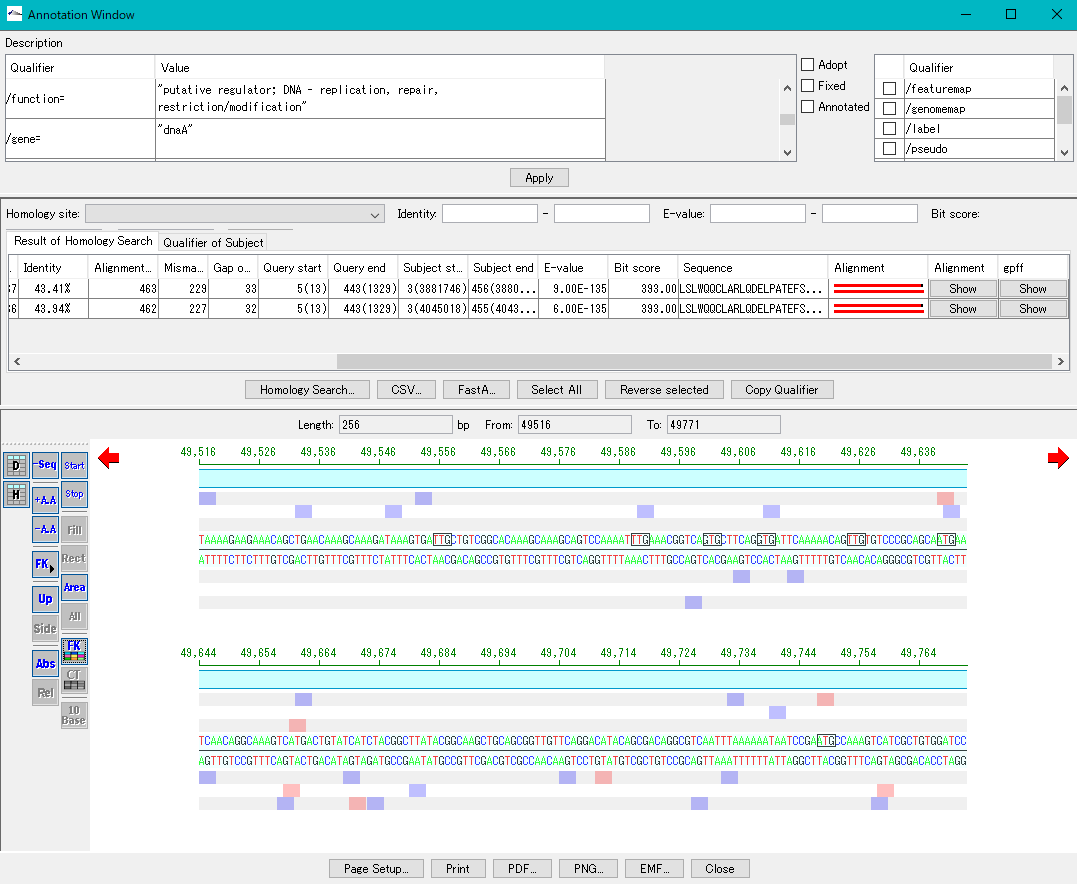
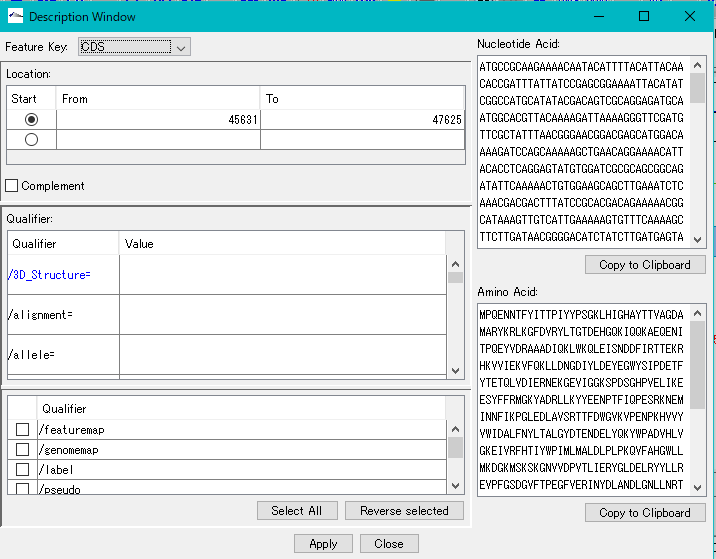
配列ビューワ アノテーションビューア
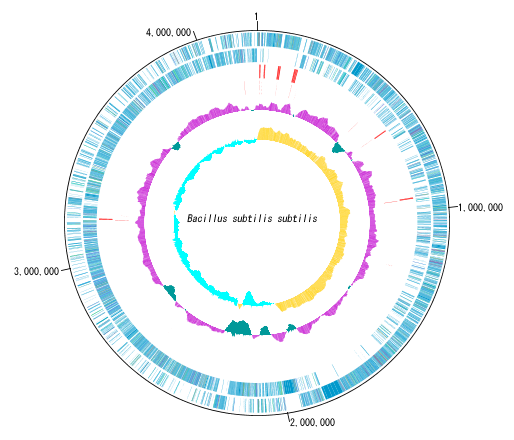
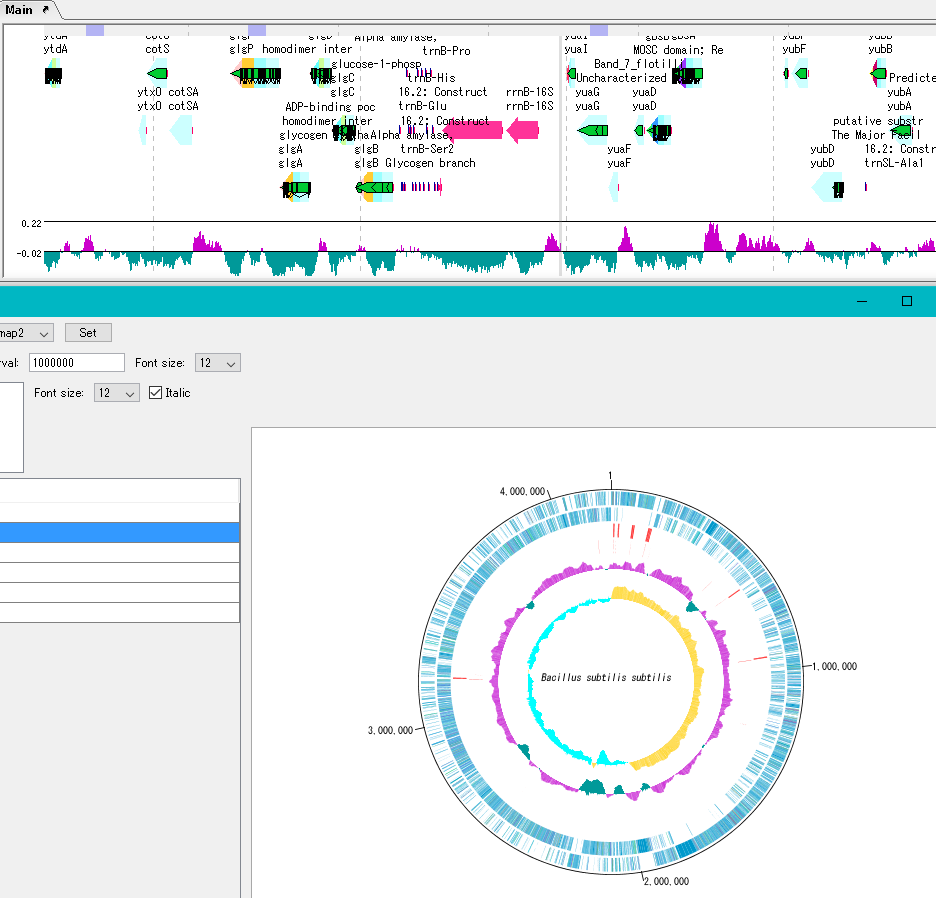
アノテーションビューア 環状ゲノムマップビューワ
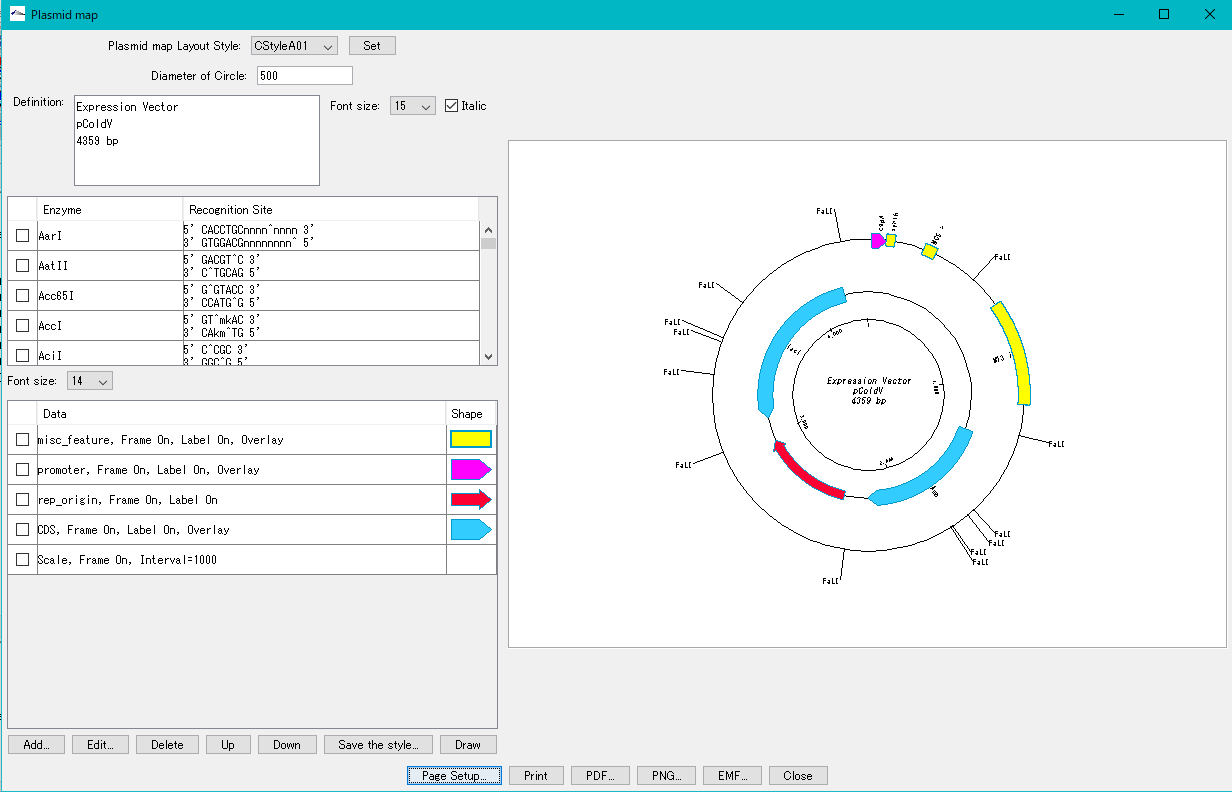
環状ゲノムマップビューワ プラスミドマップビューア
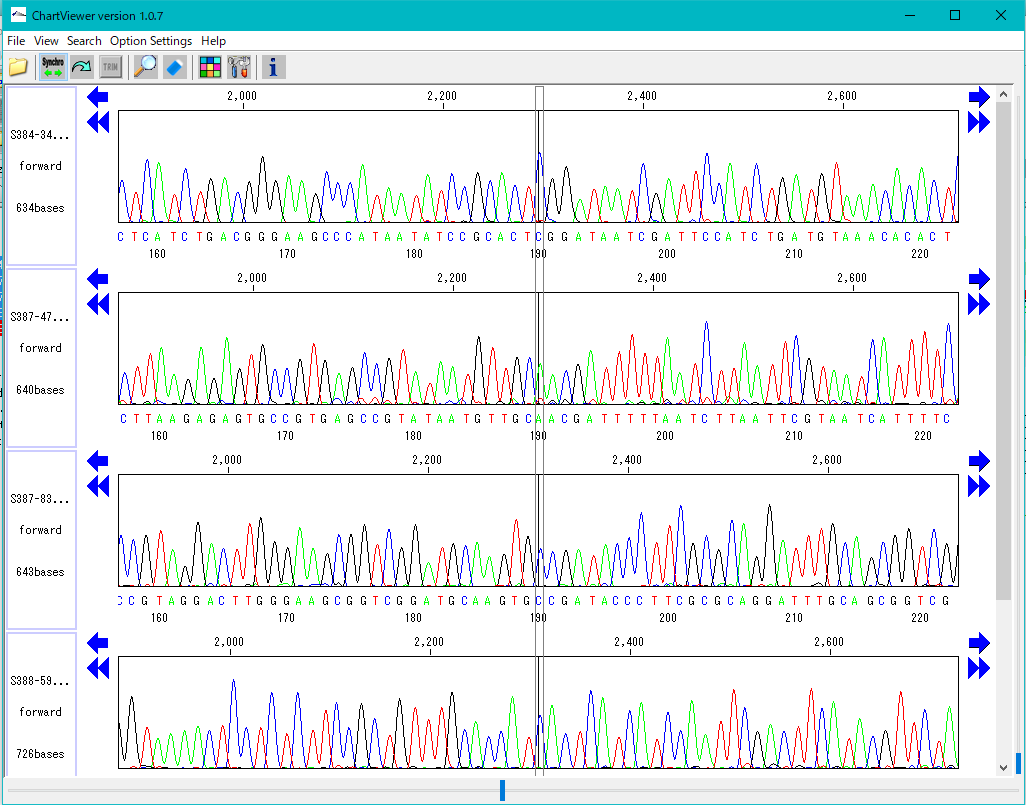
プラスミドマップビューア トレース波形ビューア
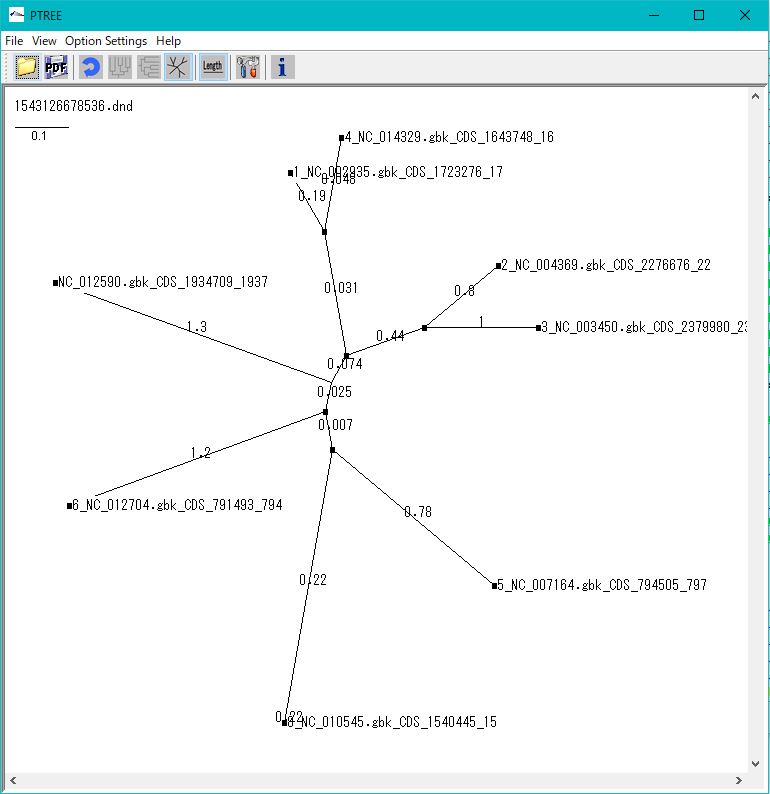
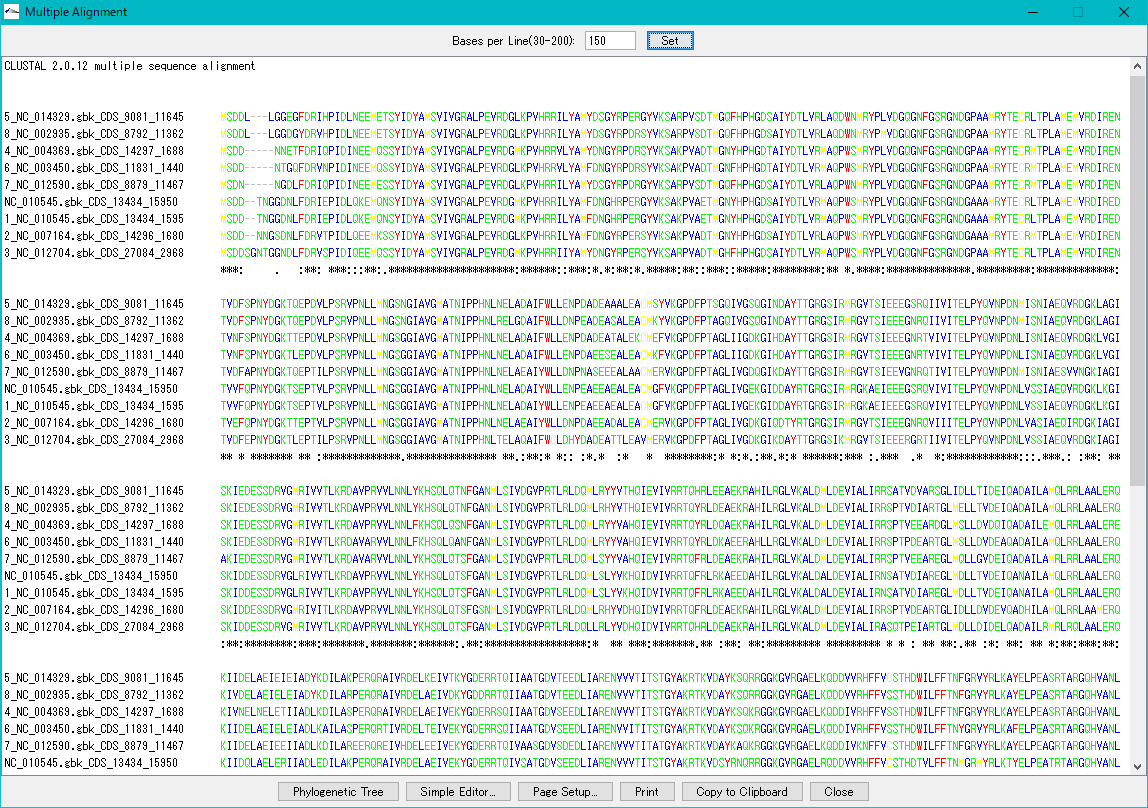
トレース波形ビューア 分子系統樹ビューア
分子系統樹ビューア フィーチャーキー検索
フィーチャーキー検索 パターン検索
パターン検索 バッチ(一括)ホモロジー検索
バッチ(一括)ホモロジー検索 プライミング部位検索
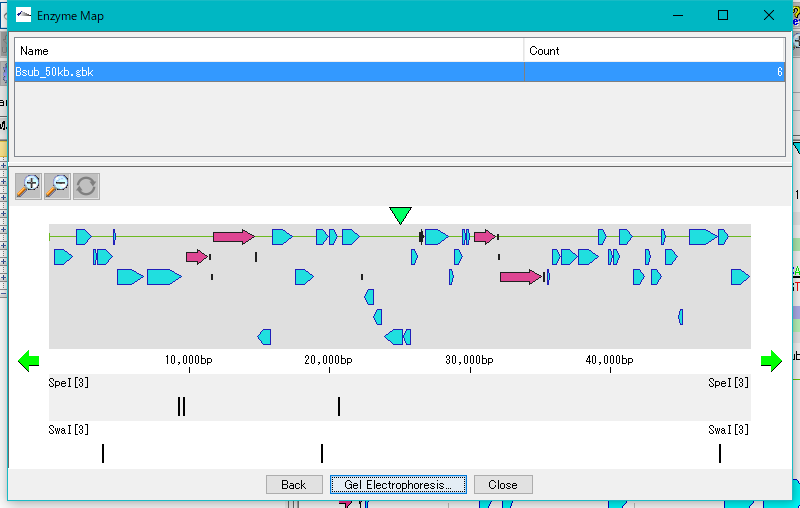
プライミング部位検索 制限酵素解析
制限酵素解析 プライマー設計
プライマー設計 コンティグブリッジ
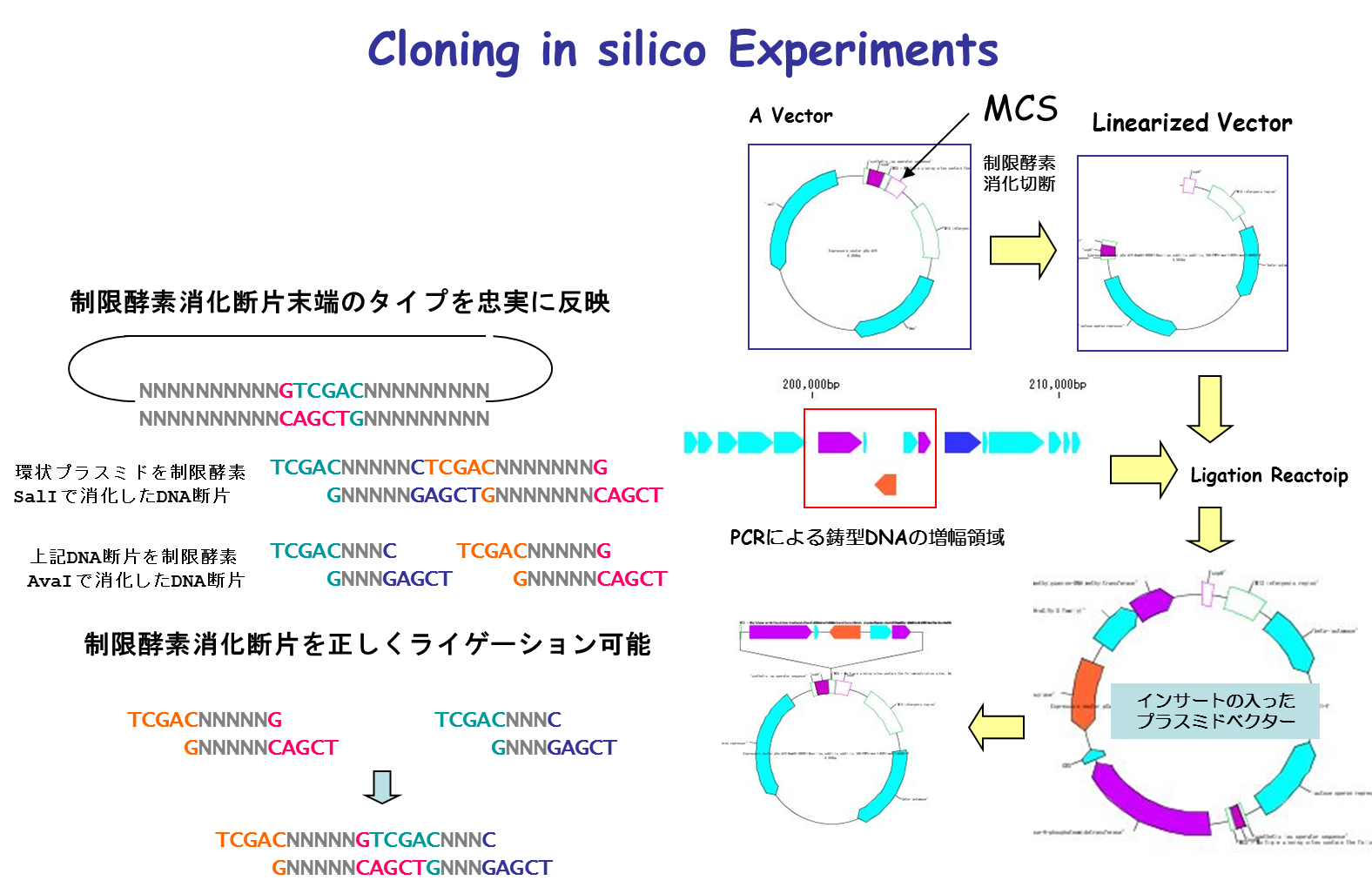
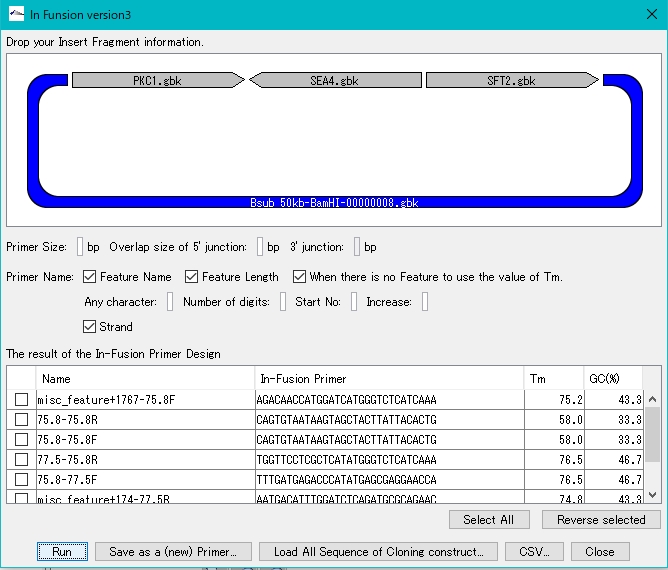
コンティグブリッジ DNA断片修飾
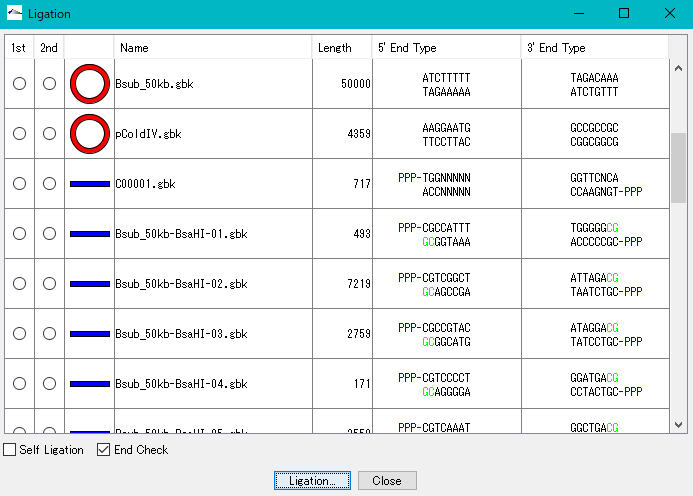
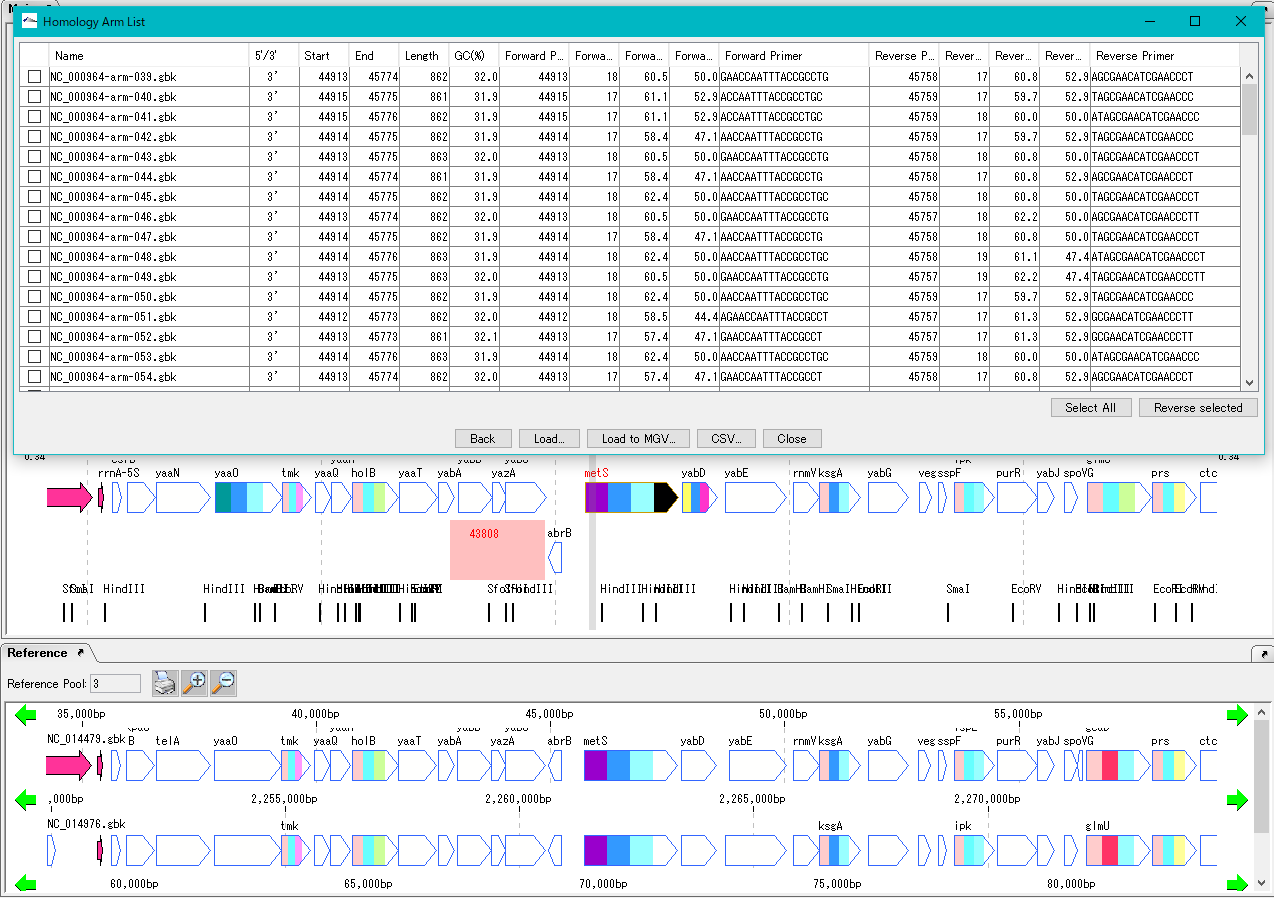
DNA断片修飾 相同組換処理
相同組換処理 DNA組成解析
DNA組成解析 コドン解析
コドン解析 ORF解析
ORF解析 配列データベースの管理
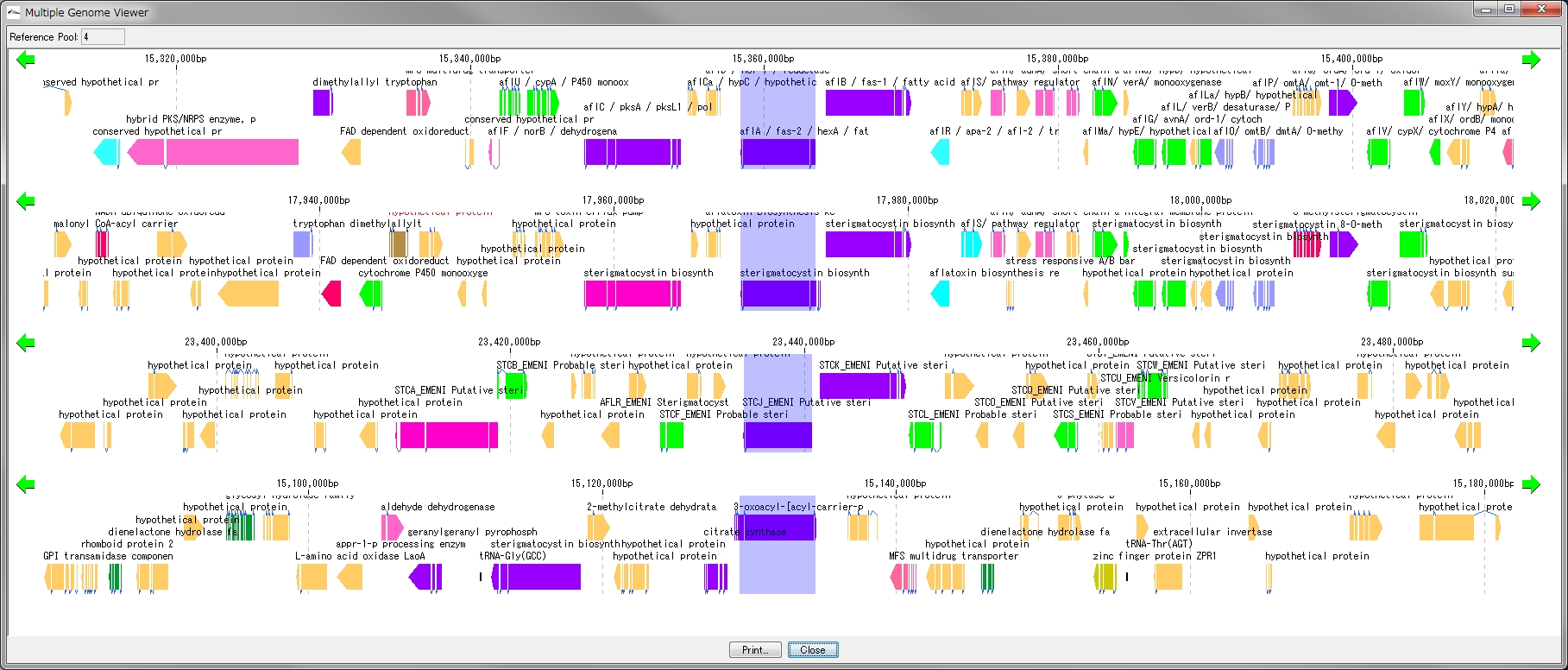
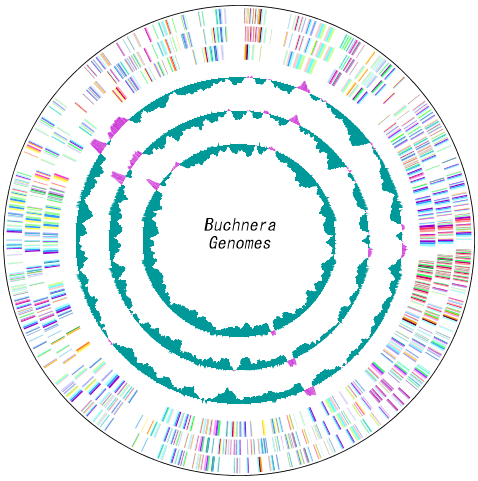
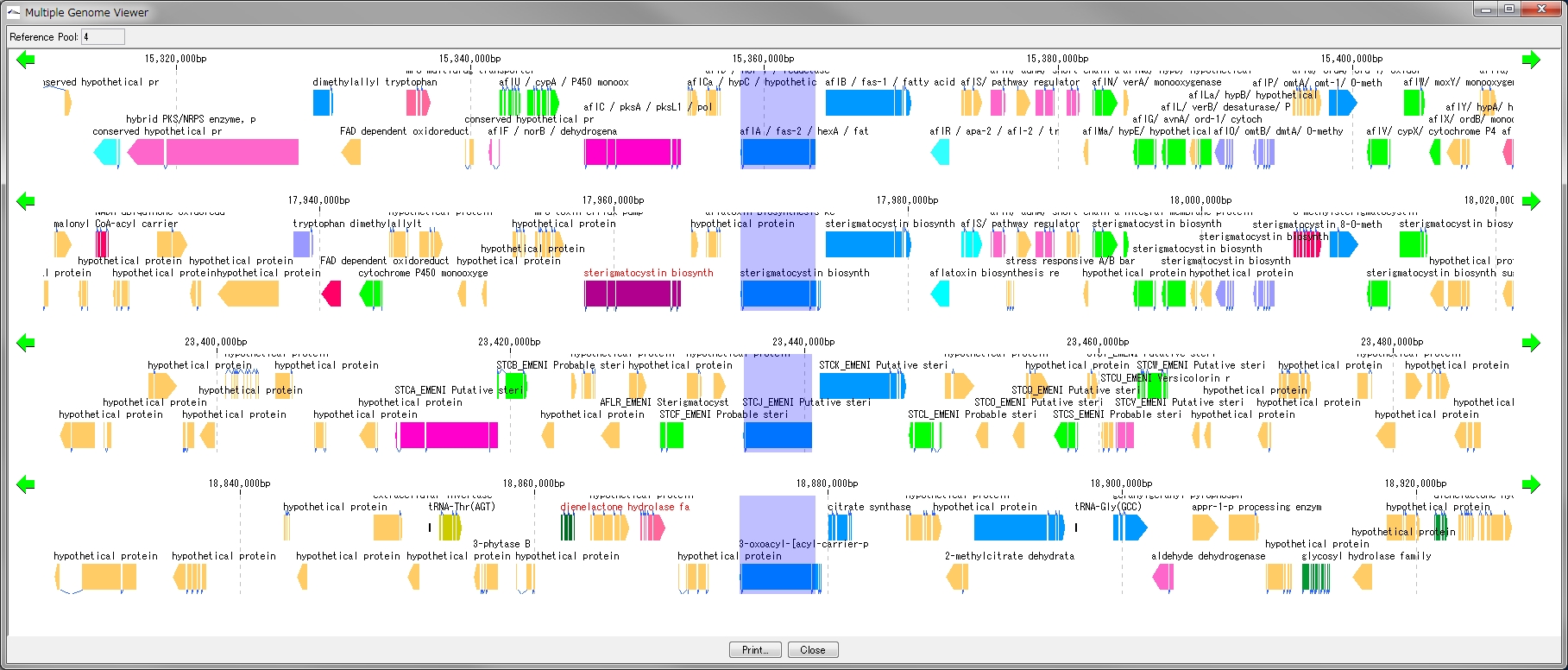
配列データベースの管理 多重環状ゲノムマップ
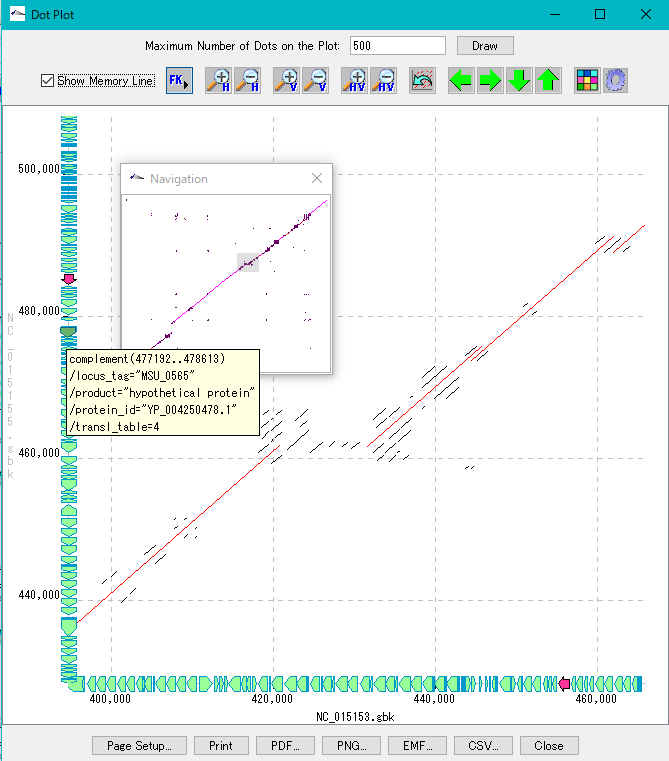
多重環状ゲノムマップ ドットプロット解析
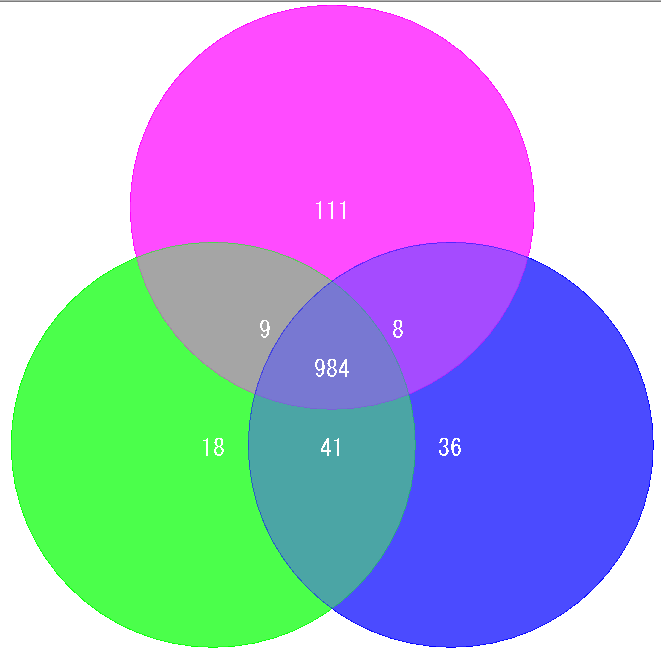
ドットプロット解析 ベンダイアグラム解析
ベンダイアグラム解析 逆相補鎖配列の生成
逆相補鎖配列の生成 設定・インストール
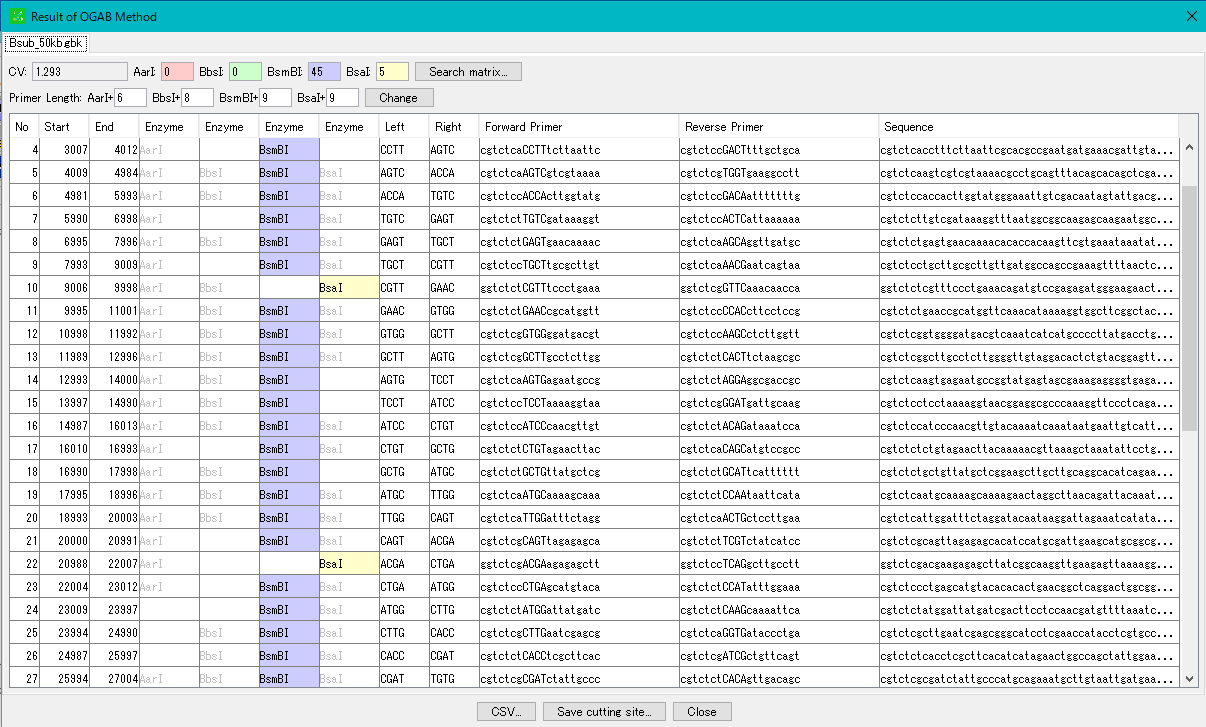
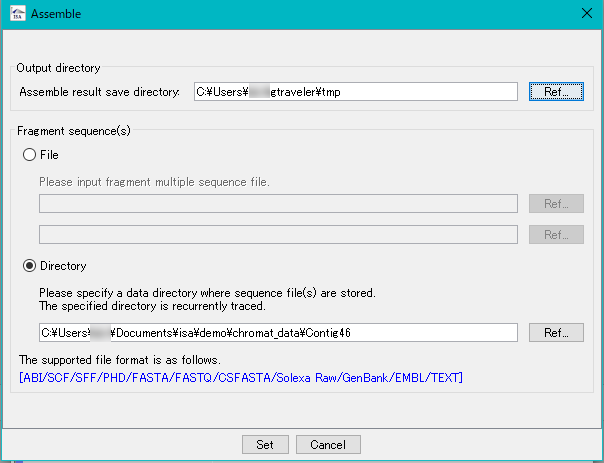
設定・インストール インシリコアセンブラー
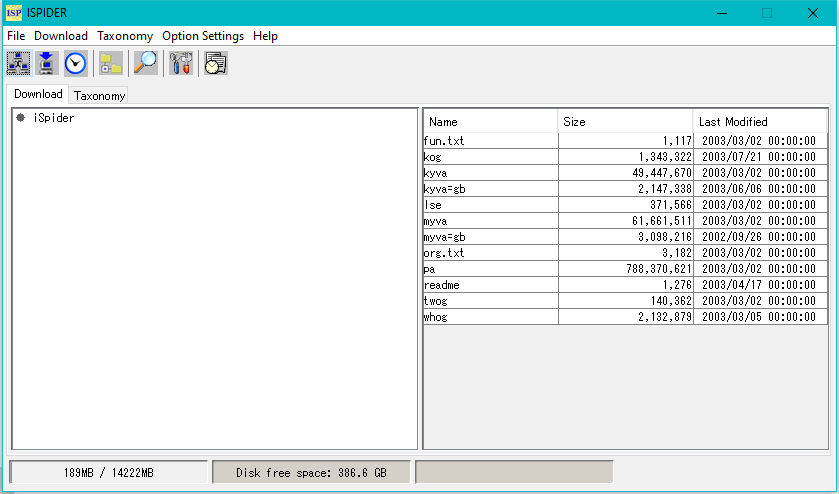
インシリコアセンブラー アイ・スパイダー(iSpider)
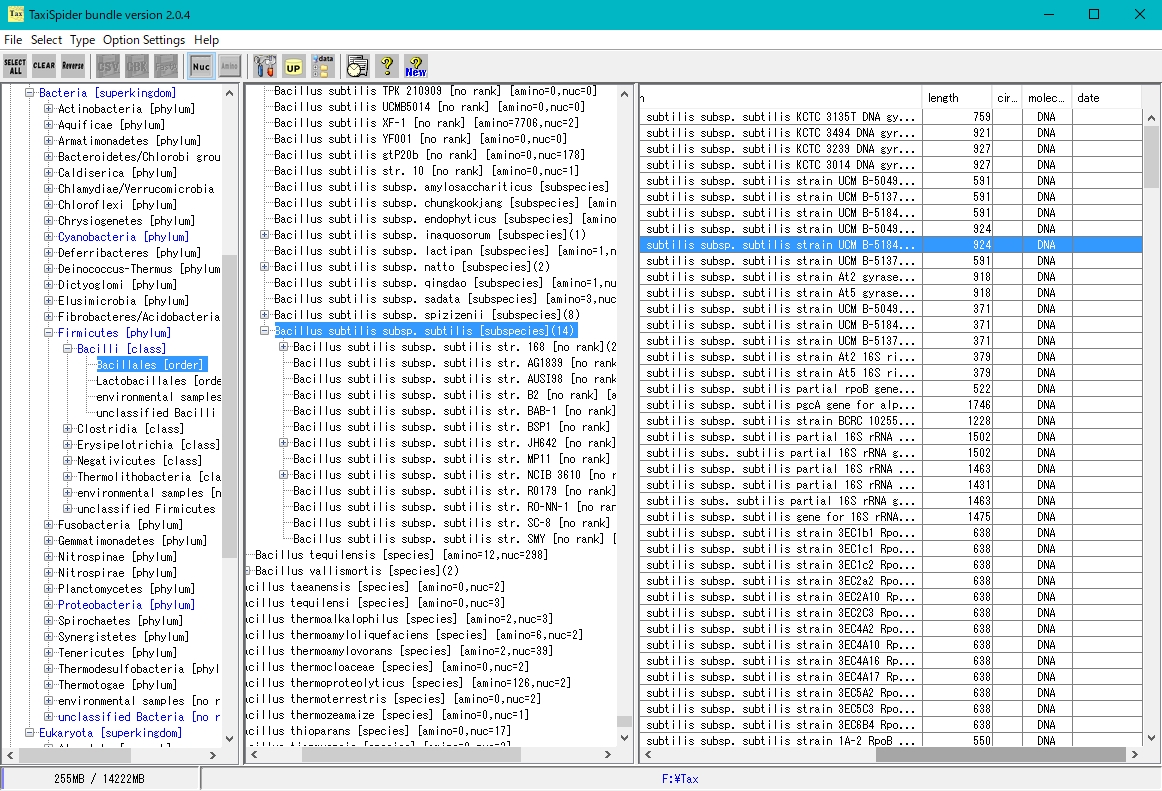
アイ・スパイダー(iSpider) タクシースパイダー (TaxiSpider)
タクシースパイダー (TaxiSpider) ARM
ARM GenBank GBFF Expander
GenBank GBFF Expander GenBankファイルチェッカー
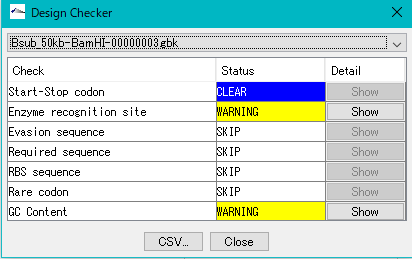
GenBankファイルチェッカー